

Sequence of shopping carts in-depth analysis with R – Clustering
Author

published
This is the second part of the in-depth sequence analysis. In the previous post, we processed data in the required format, plotted a Sankey diagram, and did some distribution, frequency, time lapse and entropy analysis with visualization. We will apply clustering in this post.
Clustering is an exploratory data analysis method aimed at finding automatically homogeneous groups or clusters in the data. It simplifies a large number of distinct sequences in a few types of trajectories.
Let’s assume that we want to identify four segments of customers based on their behavior (purchase sequences). We will use the hierarchical clustering method Ward for clustering our customers with the following code:
# CLUSTERING
library(cluster)
df.om <- seqdist(df.seq, method='OM', indel=1, sm='TRATE', with.missing=TRUE) # computing the optimal matching distances
clusterward <- agnes(df.om, diss=TRUE, method="ward") # building a Ward hierarchical clustering
df.cl4 <- cutree(clusterward, k=4) # cut the tree for creating 4 clusters
cl4.lab <- factor(df.cl4, labels=paste("Cluster", 1:4)) # creating label with the number of cluster for each customer
Once we have identified clusters, we can plot three types of graphics we are familiar with from the previous post. These graphics can help us to identify the typical patterns that characterize the clusters. We will start with a distribution analysis for each cluster which shows the state distribution at each time point (the columns of the sequence object), continue with a frequency plot, and finish with a mean time spent in each state plot:
# distribution chart
seqdplot(df.seq, group=cl4.lab, border=NA)
# frequence chart
seqfplot(df.seq, group=cl4.lab, pbarw=T, border=NA)
# mean time plot
seqmtplot(df.seq, group=cl4.lab, border=NA)
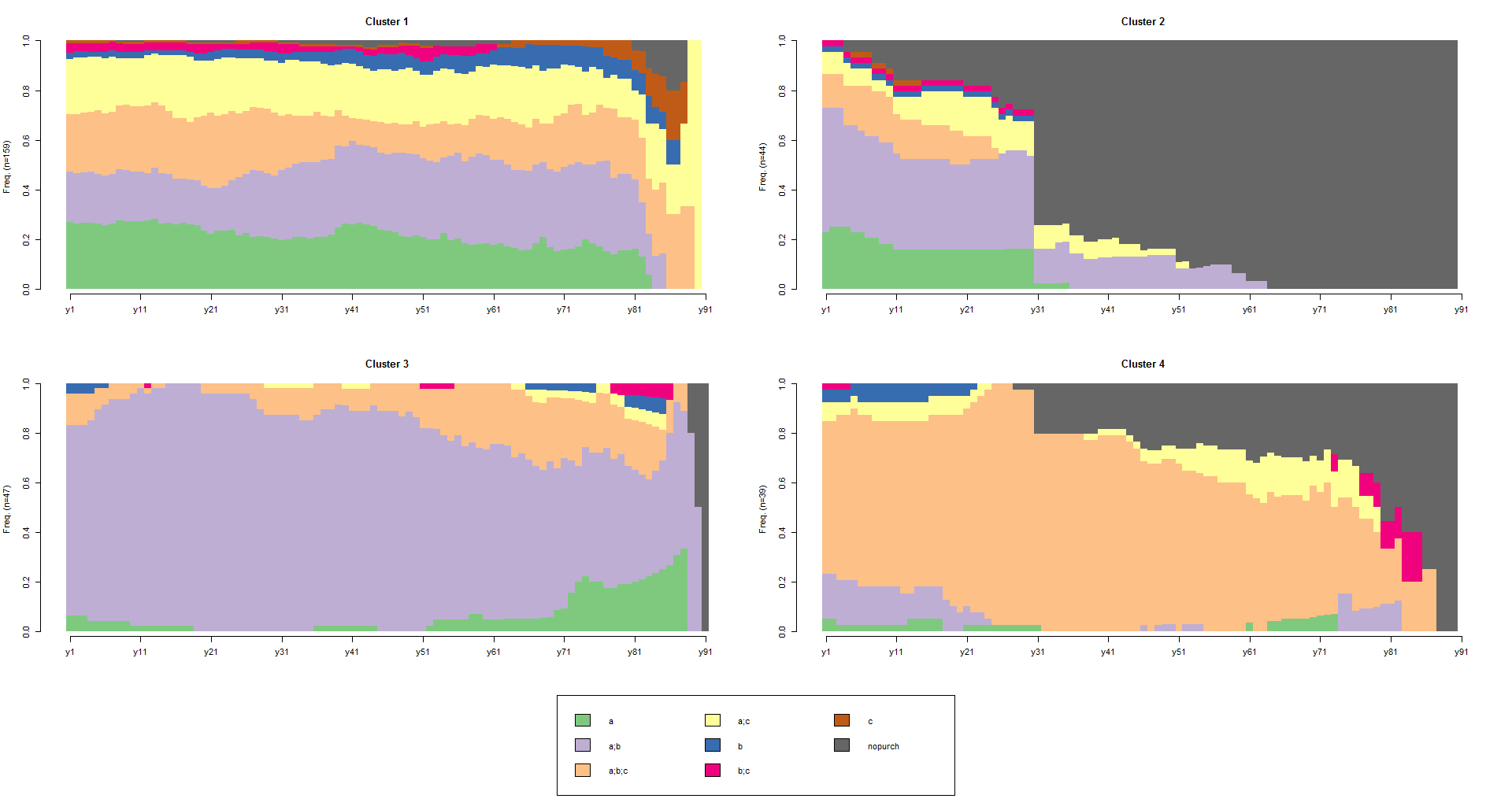

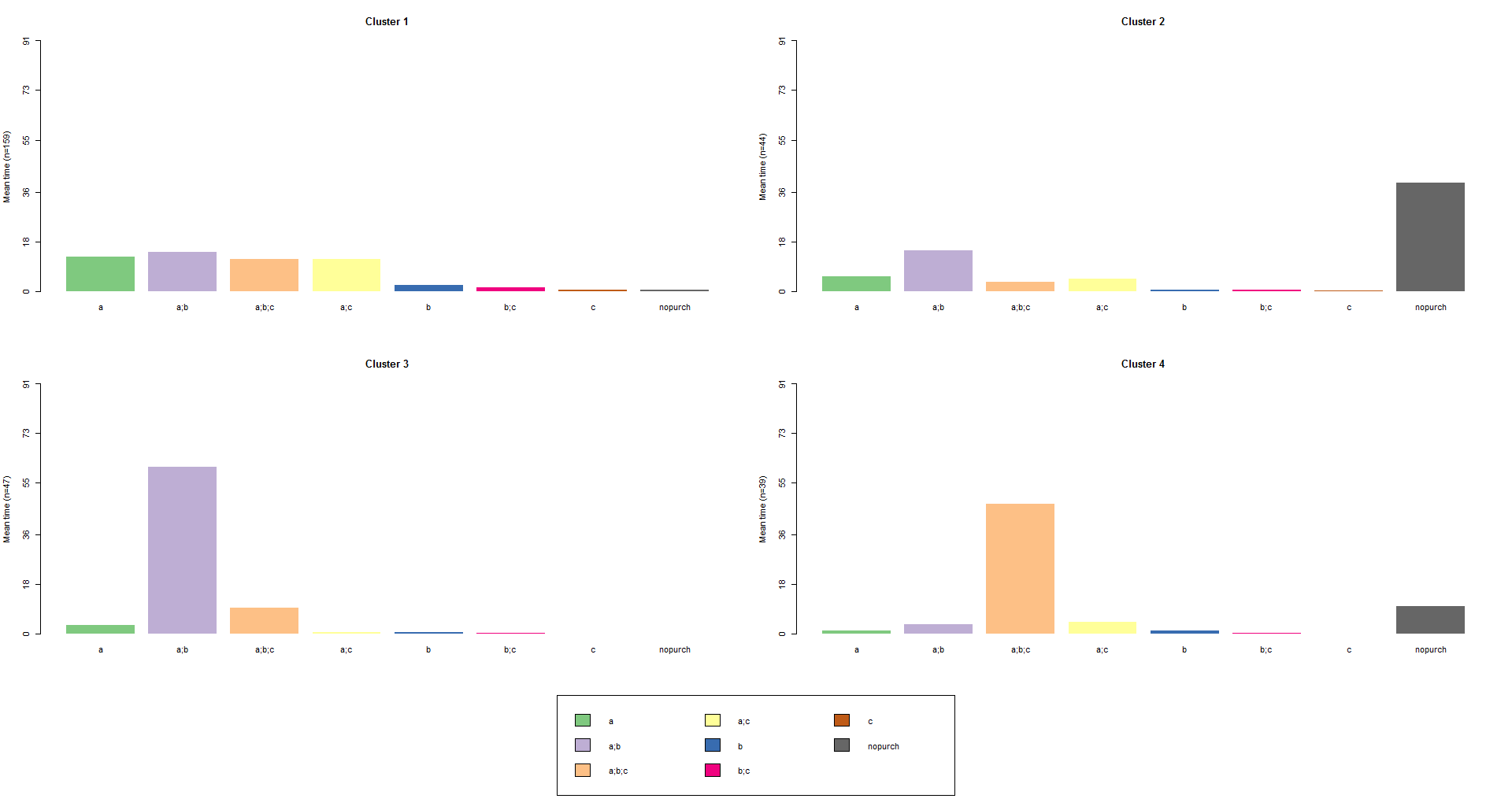
It is also possible an advanced approach of clustering. The command below finds and plots the representative set that, with a neighborhood radius of 10% (default tsim value), covers at least 35% (trep parameter) of the sequences in each of the four cl4.lab groups:
seqrplot(df.seq, group=cl4.lab, dist.matrix=df.om, trep=0.35, border=NA)
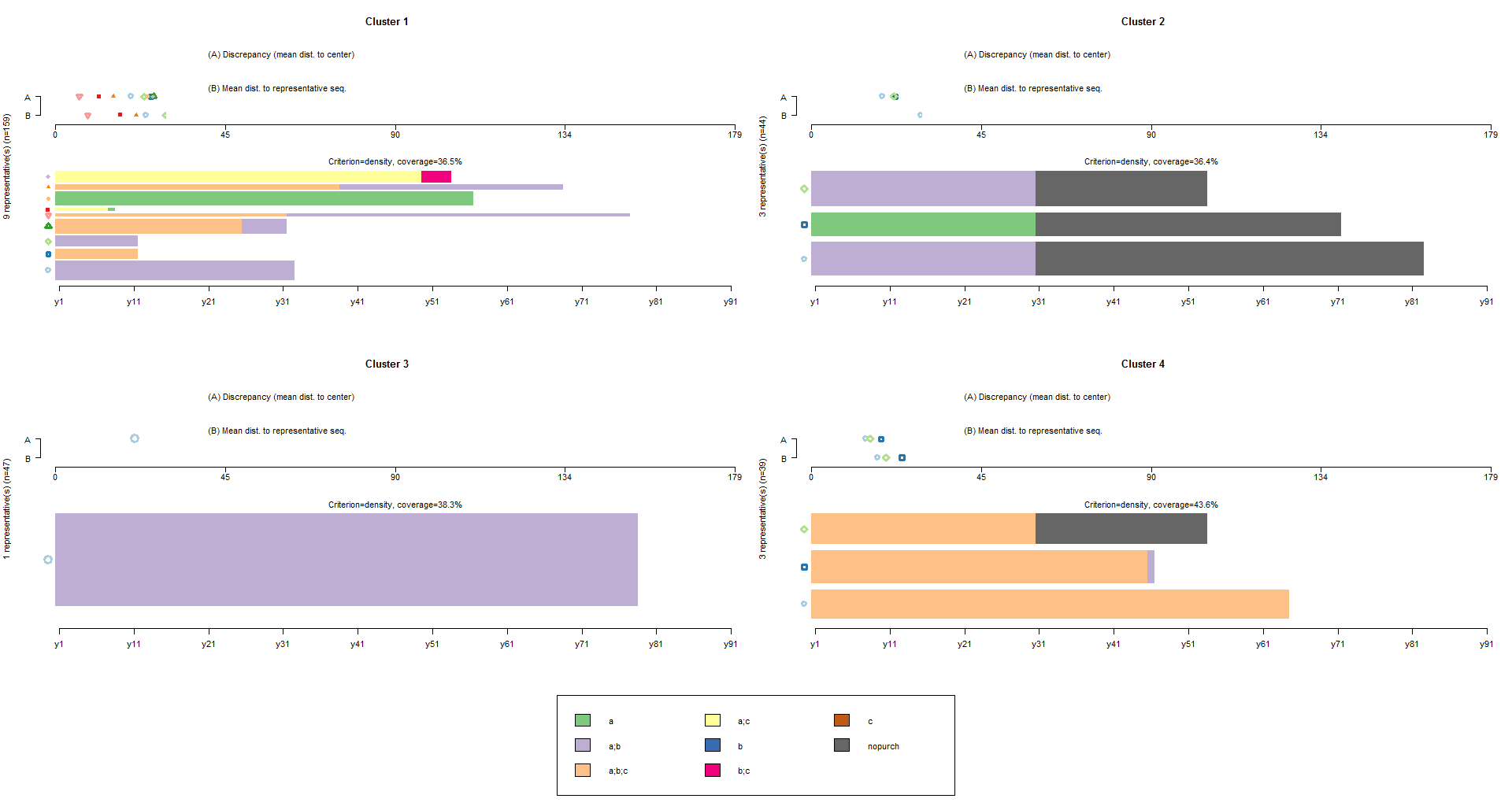
In the resulting plot the selected representative sequences are plotted bottom-up according to their representativeness score with a bar width proportional to the number of sequences assigned to them. At the top of the plot, two parallel series of symbols standing each for a representative sequence are displayed horizontally on a scale ranging from 0 to the maximal theoretical distance Dmax. The location of the symbol associated with the representative sequence indicates on axis A the discrepancy within the subset of sequences and on axis B the mean distance to the representative sequence.
We learn from the plots that nine, three, one and three representatives, respectively, are necessary for each of the four groups to achieve the 35% coverage and that the actual coverage is 36.5%, 36.4%, 38.3% and 43.6%, respectively.
So, what is the main point of preceding analysis? We can use it for:
- Defining customers who left our company using solutions I mentioned in the previous post and intending to recover them;
- Both predicting customer’s sequences and intending to affect them by recommending the right product at the right time during the customer’s life-time. The right product best matches another one and leads to increase customer’s life-time value;
- Defining unsuccessful combination of products/carts;
- Clustering customers based on their sequences and working with homogeneous segments;
and so on.
Author
